CCseqBasic --snp
~ investigate the allelic skew
The pre-requisite of a data-rich SNP-style run is a SNP of interest closer than half the --ampliconSize (default 350b) from a RE cut site (175b). Preferably the SNP of interest should be within +/- 50 bases from the RE cut site, and the closer to the cut site, the better. If all your SNPs are farther from RE cut sites than this, you cannot probe them via this method. However, note that having multiple SNPs of interest under your very capture oligo (within the very oligonucleotide you used to capture the data with) may affect your pull-down.
As this method only probes reads which overlap the SNPs of interest - the data depth is greatly reduced (the more the farther away the SNP of interest is from the closest RE cut site). Do not expect high quality interaction profile, but rather a plot from which you can "roughly determine" whether the general landscape of interactions was different between the allelic forms.
You are recommended to use --bowtie2 as a flag in your CCseqBasic SNP-specific run, as it has better tolerance for SNPs in mapping. However, the duplicate filtering of CCseqBasic struggles a little with bowtie2 multimapped reads, so best practise (at the time of writing this 01Feb2019) is to map with both --bowtie1 and --bowtie2 , and check no artifacts accumulate in bowtie2, and check that SNPs get treated fairly in bowtie1 The very best way would be to generate a N-masked genome (and its associated bowtie1 build), which replaces the SNPs of interest with Ns , and update that genome to the /conf/genomeBuildSetup.sh file. This genome can now be used in bowtie1 mapping without biasing the true SNP distribution over the sites of interest.
To run a SNP-specific run, run the pipeline normally, for example :
CCseqBasic5.sh \
-c /t1-data/user/telenius/capturesiteREfragments.txt \
-s C57captureTest \
--pf /public/telenius/CAPTUREC_DATA/C57captureTest_run4 \
--genome mm9 \
--chunkmb 1012 \
--R1 /t1-data/user/telenius/R1_001.fastq \
--R2 /t1-data/user/telenius/R2_001.fastq \
To turn on the SNP-specific mode , add flag
--snpTo yield run command like :
CCseqBasic5.sh \
-c /t1-data/user/telenius/capturesiteREfragments.txt \
-s C57captureTest \
--pf /public/telenius/CAPTUREC_DATA/C57captureTest_run4 \
--genome mm9 \
--chunkmb 1012 \
--R1 /t1-data/user/telenius/R1_001.fastq \
--R2 /t1-data/user/telenius/R2_001.fastq \
--snp
This command will read the SNP locations of interest from the last 2 columns of the capture fragment parameter file :
( these are 1-based coordinates - like f.ex. gff and sam files, not 0-based like f.ex. bed files. In 1-based coordinates a single base is defined by for example chr1 1 1 (chr1 region from the 1st base to the 1st base - i.e. the 1st base itself) I.e. in 1-based coordinates only ONE coordinate is needed to define a SNP location, instead of both start and end coordinates. More about 1-based and 0-based files f.ex. in here )
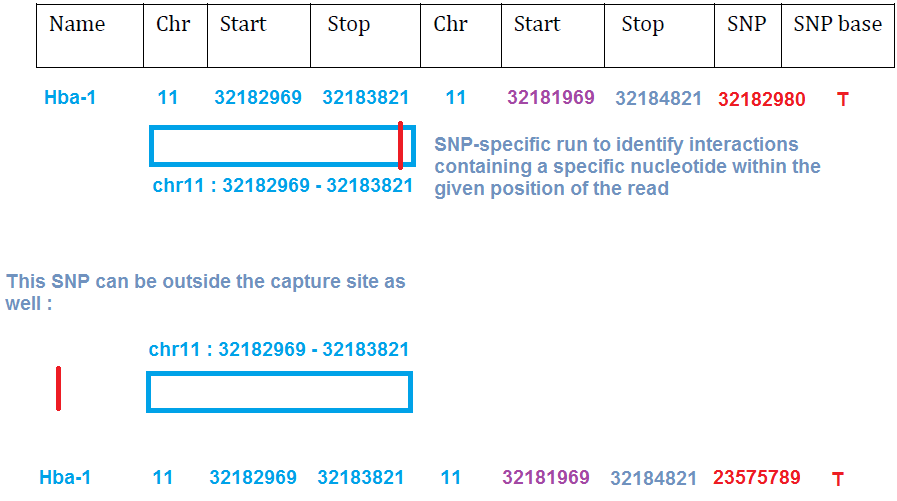
I.e. to make the run SNP-specific, we need to add in the 8th and 9th column, to describe the desired SNP, and the desired allele, like so :
Oct-4 17 35641755 35643135 17 35640755 35644135 35641789 G Pkdrej 15 85651837 85652458 15 85650837 85653458 85652165 A
Even if you only have SNPs of interest in some of your capture sites, you should include all of your capture sites to your capture fragment parameter file, to enable the run properly filter the ambiguous interactions which are assigned to more than one capture site. To exclude these "only for filtering" capture sites from the output reports and visualisations, give "dummy coordinates" 1 A as the last columns of the capture fragment parameter file, like so :
TP53 11 69392395 69394473 11 69391395 69395473 1 A
Each SNP-specific file can contain each capture site ONLY ONCE ( if you want to run for multiple alleles of the base of interest, you need to run several runs ).
For example, to probe both major alleles of the above example Oct-4 (G,A) and Pkdrej (A,G), we need 2 runs like so :
Oct-4 17 35641755 35643135 17 35640755 35644135 35641789 G Pkdrej 15 85651837 85652458 15 85650837 85653458 85652165 A TP53 11 69392395 69394473 11 69391395 69395473 1 Aand
Oct-4 17 35641755 35643135 17 35640755 35644135 35641789 A Pkdrej 15 85651837 85652458 15 85650837 85653458 85652165 G TP53 11 69392395 69394473 11 69391395 69395473 1 A
Details about the capture site parameter file, its file format, and editing instructions available here : fragmentfile.html
Walk through 'vignette' using the data set of original CaptureC method publication : walkthrough