CCseqBasic
~ Interaction counters in F2 / F3 / F5 / F6 folders

If you haven't yet red the "analysis steps" and "output folders" sections of the CCseqBasic main page , this is a good time to do so :
- if you don't know what the F2 / F3 / F5 / F6 folders are, and what are the analysis steps performed in each of them, interpreting the report counters is going to be error-prone !
Here more details and illustrations
about assigning fragments to "captures" and "reporters", like this :
 ( i.e. marking the mapped fragments to reflect the true results of the NG-CaptureC experiment )
( i.e. marking the mapped fragments to reflect the true results of the NG-CaptureC experiment )
Before reading the text below , check the link above : browse through the illustrations - to get a clear picture what the text below refers to !
1a Actual reported fragments : 182 1b Actual reported CIS fragments : 88 1c Actual reported TRANS fragments : 94 Hba-1 1 Capture fragments (final count): 150 Hba-1 3a Reporter fragments (final count) : 110 Hba-1 3b Reporter fragments CIS (final count) : 46 Hba-1 3c Reporter fragments TRANS (final count) : 64 Hba-2 1 Capture fragments (final count): 102 Hba-2 3a Reporter fragments (final count) : 72 Hba-2 3b Reporter fragments CIS (final count) : 42 Hba-2 3c Reporter fragments TRANS (final count) : 30explaining the above :
Total capture counts :
1a Actual reported fragments : All reported fragments (CIS+TRANS), all capture sites. 1b Actual reported CIS fragments : All reported fragments (CIS), all capture sites. 1c Actual reported TRANS fragments : All reported fragments (TRANS), all capture sites.
Capture-site-wise counters (here two capture sites Hba-1 and Hba-2)
Hba-1 1 Capture fragments (final count): All capture fragments, Hba-1 capture site.
This many fragments which mapped to the CAPTURE-site of Hba-1.
The below 3 counters are then the REPORTERS which were found in
the reads containing one or more of these CAPTURE fragments.
Hba-1 3a Reporter fragments (final count) : All reported fragments (CIS+TRANS),
interacting with Hba-1 capture site.
Hba-1 3b Reporter fragments CIS (final count) : All reported fragments (CIS),
interacting with Hba-1 capture site.
Hba-1 3c Reporter fragments TRANS (final count) : All reported fragments (TRANS),
interacting with Hba-1 capture site.
Hba-2 1 Capture fragments (final count): Hba-2 capture-site RE-fragment mapping read-fragments
(one or more per read)
Hba-2 3a Reporter fragments (final count) : All reported fragments (CIS+TRANS),
interacting with Hba-2 capture site.
Hba-2 3b Reporter fragments CIS (final count) : All reported fragments (CIS),
interacting with Hba-2 capture site.
Hba-2 3c Reporter fragments TRANS (final count) : All reported fragments (TRANS),
interacting with Hba-2 capture site.
The above counts are not "normal" in their distribution ( they originate from a TESTER file, which is not realistic )
explaining the above :
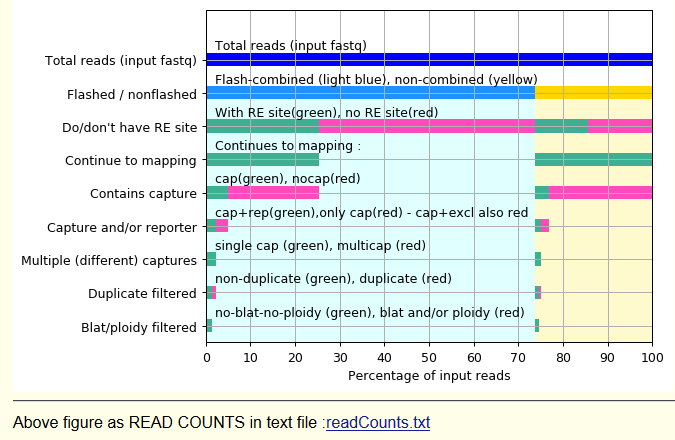
Total reads (input fastq) Read count of all reads - as 100%
Flashed / nonflashed Flash-combined reads in grey (from this point downwards)
Non-combined reads in purple (from this point downwards)
If you sequenced 150+150 bases PE reads, and sonicated to ~300 bases fragments,
you should have most reads "flashed".
If you sequenced 75+75 bases PE reads, and sonicated to ~300 bases fragments,
you have less reads "flashed, but still many.
If you sequenced 40+40 bases PE reads, and sonicated to ~300 bases fragments,
you have almost all reads "nonflashed".
Do/don't have RE site
green : the desired RE-site can be seen at least once within the R1+R2 sequence of the read
red : the desired RE-site can NOT be seen at least once within the R1+R2 sequence of the read
Note : the non-flashed reads may have the RE site "invisible" - i.e. between the two halves of the read.
Continue to mapping
All non-flashed reads (purple) continue to mapping (stay green).
Of the flashed reads (grey) we filter the ones where we didn't see the desired RE-cut sequence
( we already know these cannot have a ligation product )
Contains capture
has capture (green) : the reads where some part(s) mapped to the desired captured RE fragments
no capture (red) : the reads where no part(s) mapped to the desired captured RE fragments
Contains capture and/or reporter
has capture reporter (green) : the reads where also "another fragment" was seen.
no "capture+reporter" (red) : the reads where 1) no another fragment, or
2) only too-close-to-capture-site fragment ("exclusion fragment") was seen
The above counters will help you to differentiate between these situations :
1) Wrongly given RE-coordinates in CCseqBasic run
(all reads mysteriously vanish when asked do they "contain capture site")
2) Capture-C experiment had a weak capture step (
signal coming from genomic background - not actually enriched for captured sites)
To confirm this, it may be necessary to visualise the bam file of F1 folder as well
- if the not-analysed reads show just genomic background
( no higher-than-background signal in the capture sites ), that confirms this diagnosis.
3) Capture-C experiment had a poor ligation step (only single fragments going to sequencng)
4) Capture-C experiment had a poor digestion step
(the RE-site containing reads only map to capture sites, and don't report anything)
5) Repeat-rich capture oligo design (filtered heavily in blat-filtering step)
6) Library not sequenced to exhaustion (only very few duplicates seen)
7) Capture sites within a single interaction domain (interacting with each others as well)
- a lot of "multicapture" reads (which cannot be interpreted)
It is generally not recommended to have interacting regions in same design
(f.ex. enhancers and promoters in same design)
The same counts as numbers instead of percentages :
all=86320 All read pairs
allflashed=3001 All flashed read pairs
allnonflashed=83319 All non-flashed read pairs
REflashed=2893 RE-cut site containing flashed read pairs
REnonflashed=157924 RE-cut site containing non-flashed read pairs
continuesToMappingFlashed=2893 Flashed read pairs continuing to mapping (only the ones having RE cut site)
continuesToMappingNonflashed=83313 Non-flashed read pairs continuing to mapping (all reads)
containsCaptureFlashed=1434 After mapping, read is seen to contain a capture site
containsCaptureNonflashed=78383
containsCapAndRepFlashed=104 After mapping, read is seen to contain also an interaction fragment
containsCapAndRepNonflashed=73588
singleCapFlashed=87 Only one capture site seen within the read pair
singleCapNonflashed=50027 (no f.ex enhancer-promoter-pairs)
multiCapFlashed=17 Multiple capture sites seen within the read pair
multiCapNonflashed=23561 (design rich in f.ex enhancer-promoter-pairs)
nonduplicateFlashed=86 Unique reads - flashed (not duplicates)
nonduplicateNonflashed=47480 Unique reads - nonflashed (not duplicates)
blatploidyFlashed=0 Fragments filtered as blacklisted or homology regions - flashed
blatploidyNonflashed=47480 Fragments filtered as blacklisted or homology regions - nonflashed
Interaction counters in F2 / F3 / F5 / F6 folders
In general these counters are very straightforward :
The code more or less reports "every step of the way".
So - all the numbers correspond to CHRONOLOGICAL steps during the analysis.
The only exception is number 6 (duplicate reads) - which comes way too early.
All the other numbers correspond to the filtering order in the script.
####################################################
We have divided each READ into RE-cut FRAGMENTS - and we count them in various ways
i.e. counters (1-10)
###################################################
So, first we have all fragments (1), and count them in various ways (1-10)
01 Number of capture sites loaded: 16
02 Restriction enzyme fragments loaded: 6199203
03 Lines in sam file header: 23
04 Data lines in sam file: 59265143
06 Unmapped fragments in SAM file: 2851157
06c Duplicate reads: 9801446
07 Mapped fragments: 56413986
09 Proximity exclusion fragments (Pre PCR duplicate removal): 2101978
10 Reporter fragments (Pre PCR duplicate removal): 10374399
####################################################
We reconstruct the READS from the RE-cut FRAGMENTS - and we count them in various ways
i.e. counters (11-15)
###################################################
After mapping in bowtie, our reads are in the file so, that all fragments of one "read" are one after another.
Then, once we know we have all the fragments of a read, we look at them,
and if we didn't lose all our reads in filtering steps above (counters 1-10)
We further filter the reads.
Read entering further analysis stages have to have
1) a capture fragment
2) reporter fragment
but MAY STILL contain multiple different captures
We also count the FRAGMENTS within the reads, which had at least one mapped fragment (counter 13)
####################################################
Detailed counts of fragments and their composition (before duplicate-filtering)
i.e. counters (11e,11ee)
###################################################
Before duplicate filtering your "preliminary counts" of reporters are in 11e and 11ee
How to read these lines :
11ee Total number of reads having captures in composition Hom9:1 (1)
, having 1 reporters and 0 exclusion fragments : 918274
11ee Total number of reads having captures in composition Hom9:1 Pam6:1 (2)
, having 1 reporters and 0 exclusion fragments : 3
11ee Total number of reads having captures in composition Hom9:2 (3)
, having 1 reporters and 0 exclusion fragments : 255796
The first line (1) means :
Hom9 capture. Capture fragments seen : 1.
Reporter fragments seen 1. Exclusion fragments seen 0.
Total count of these : 918274
Second line (2) :
Hom9 capture, having also Pam6 capture.
Capture fragments seen : 1 (in Hom9), Capture fragments seen : 1 (in Pam6).
Reporter fragments seen 1. Exclusion fragments seen 0.
Total count of these : 3 (so very rare)
Third line (3) :
Hom9 capture. Capture fragments seen : 2. (so, Hom9 capture was seen in 2 fragments of the read)
Reporter fragments seen 1. Exclusion fragments seen 0.
Total count of these : 918274
####################################################
We duplicate filter our reads
i.e. counters (12-16)
###################################################
Then the code makes duplicate filter (counters 16).
So - we get rid of reads which most probably are each other's PCR duplicates.
The "composition" is reported the same way as 11e and others above.
Now we still continue reporting "global statistics" after the duplicate filtering,
and continue all the way upto number 25.
12 Total number of reads entering duplicate-filtering - should be same count as 11f : 9886207
13 Count of fragments in Reads having at least one informative fragment : 21410249
14a Reads having 2 fragments: 8291768
14a Reads having 3 fragments: 1551218
14a Reads having 4 fragments: 43048
14a Reads having 5 fragments: 171
14a Reads having 6 fragments: 2
14b Reads having 2 informative fragments: 8291768
14b Reads having 3 informative fragments: 1551218
14b Reads having 4 informative fragments: 43048
14b Reads having 5 informative fragments: 171
14b Reads having 6 informative fragments: 2
16 Non-duplicated reads: 84761
####################################################
We count our duplicate filtered reads in various ways
i.e. counters (16-23)
###################################################
16b and 16bb are counters after duplicate filter.
Interpret these like the above counters 11e and 11ee
16c Proximity exclusion fragments (After PCR duplicate removal): 1191
16d Reporter fragments (After PCR duplicate removal): 92067
16f Total fragment count (after PCR duplicate removal): 84761
16g Reads having 2 informative fragments (after PCR duplicate whole-read removal): 52924
16g Reads having 3 informative fragments (after PCR duplicate whole-read removal): 29677
16g Reads having 4 informative fragments (after PCR duplicate whole-read removal): 2094
16g Reads having 5 informative fragments (after PCR duplicate whole-read removal): 64
16g Reads having 6 informative fragments (after PCR duplicate whole-read removal): 2
23 Reporters before final filtering steps 92067
####################################################
We do final filtering to the reads
i.e. counters (23-25)
###################################################
The "last filtering steps" the report mentions is :
- if WITHIN the same read one reports SAME reporter fragment twice, it is counted only once.
- if the reporter fragment is mapped so, that one end of it is in one DpnII fragment,
and other end is in other DpnII fragment
( so that the mapped reporter fragment OVERLAPS DpnII cut site), it is filtered out,
as it is believed to be mismapped read.
23 Reporters before final filtering steps 92067
24 Duplicate reporters (duplicate-excluded if stringent was on) 40776
25 Reporter fragments reporting the same RE fragment within a single read (duplicate-excluded) 3708
25e Error in Reporter fragment assignment to in silico digested genome (see 24ee for details) 440
25ee Binary search error - fragment overlapping multiple restriction sites: 440
26 Actual reported fragments : 87919
Counters with reporters 110329
####################################################
The FINAL COUNTS (the "most important counters" )
###################################################
Now as we reach the FINAL counts for everything,
we divide to CAPTURE-SITE specific statistics.
So - basically the same counts as the above ones, but divided by CAPTURE SITE.
These report lines START with the capture site name, followed by numbers 12-17
So, these 12-17 have nothing to do with the 12-17 described above.
These are the FINAL counts for each capture site.
So basically after filtering duplicates we count the situation JUST BEFORE the duplicate filtering,
and just after.
These are the most important counters :
Olig_aGlobin 12 Reporters before final filtering steps 752513
Olig_aGlobin 13 Duplicate reporters (duplicate-excluded if stringent was on) 720951
Olig_aGlobin 14 Reporter fragments reporting the same RE fragment within a single read
(duplicate-excluded) 783
Olig_aGlobin 14e Error in Reporter fragment assignment to in silico digested genome
(see 25ee for details) 1244
Olig_aGlobin 15 Capture fragments (final count): 728013
Olig_aGlobin 16 Proximity exclusions (final count): 2538
Olig_aGlobin 17 Reporter fragments (final count) : 750486
The one you are really interested is number 17 : the actual count of the interactions to other fragments.
Usually you can only interpret the data if you have more than 30 000 fragments for each capture
in counter (17) here